
L’editing del genoma applicato sul primo paziente italiano affetto da talassemia al Bambino Gesù di Roma. La nuova terapia che offre speranze.

Un altro risultato importantissimo è stato raggiunto nella sperimentazione clinica dall’Ospedale Bambino Gesù di Roma, eccellenza nelle cure in campo pediatrico e non solo.
LEGGI ANCHE: MALATTIA DEL NEUROSVILUPPO SCOPERTA AL BAMBINO GESÙ: SOLO 7 BAMBINI AL MONDO
L’ospedale della Santa Sede ha applicato con successo una terapia sperimentale per il trattamento delle malattie genetiche del sangue, basata sul sistema dell’editing del genoma. La terapia è stata applicata su un paziente italiano, il primo ad essere trattato con questa tecnica innovativa.
Editing del genoma su primo paziente italiano al Bambino Gesù
L’Ospedale Bambino Gesù partecipa insieme ad altri 13 centri clinici in Europa, Canada e Stati Uniti a una sperimentazione di editing del genoma nella cura della talassemia e dell’anemia falciforme nei giovani adulti e adolescenti, avviata nel 2019 da Vertex Pharmaceuticals e Crispr Therapeutics. Sono stati programmati due trial clinici internazionali, che coinvolgono 45 giovani con talassemia e 45 con anemia falciforme.
Il Bambino Gesù è l’unico ospedale italiano che partecipa alla sperimentazione, con il comitato scientifico internazionale coordinato dal prof. Franco Locatelli, direttore del Dipartimento di Oncoematologia e Terapia Cellulare e Genica dell’ospedale.
Conosciamo già il prof. Locatelli per la terapia genica con cellule CAR-T. Come per quest’ultima, anche nella terapia di editing del genoma si modificano le cellule prelevate dal paziente che gli vengono successivamente inoculate con la modifica che migliora le sue condizioni.
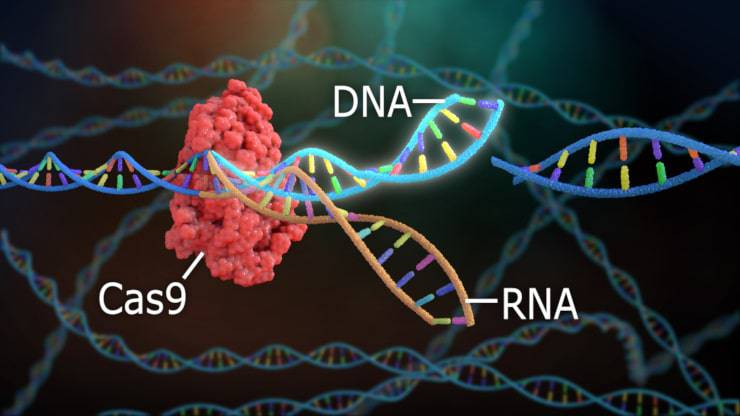
L’editing del genoma viene eseguito con il sistema CRISPR-Cas9, una tecnologia innovativa che funziona come un “correttore” del DNA ad altissima precisione. Chiamata anche “forbice genetica“, questa tecnologia è stata messa a punto dalle scienziate Emmanuelle Charpentier e Jennifer A. Doudna, che per il loro lavoro hanno ricevuto il Premio Nobel per la chimica 2020.
Il metodo si basa sull’utilizzo della proteina Cas9, una sorta di forbice molecolare che viene programmata per tagliare o modificare specifiche sequenze del DNA di una cellula. In questo modo si possono correggere diverse malattie con difetto genetico.
Il sistema CRISPR-Cas9 è un complesso di molecole biologiche formato da frammenti di RNA (acido ribonucleico) e da proteine. Il segmento di RNA è la bussola che indica il bersaglio da colpire alla proteina Cas9, che esegue il taglio o la modifica del DNA.
LEGGI ANCHE: TRAPIANTO SU ALESSANDRO MARIA MONTRESOR: IL BAMBINO SARÀ DIMESSO DALL’OSPEDALE
La terapia con editing genetico
Il trattamento terapeutico si svolge con i seguenti passaggi:
- dal paziente vengono prelevate delle cellule,
- queste cellule vengono modificate o “corrette” in laboratorio con il sistema CRISPR-Cas9,
- le cellule modificate vengono infuse nell’organismo del paziente dove si riproducono al posto di quelle difettose.
In questo modo, è possibile curare malattie come l’anemia mediterranea o talassemia e l’anemia calciforme, senza che siano necessari ulteriori interventi. Nei casi delle forme più gravi di talassemia significa non doversi più sottoporre a trasfusioni periodiche di sangue.
L’eccezionalità della notizia, oltre che nell’applicazione di questo sistema innovativo di cura, è soprattutto nella sua applicazione al primo paziente italiano, un giovane affetto da talassemia.
L’editing genetico in queste malattie del sangue avviene intervenendo sull’emoglobina. Infatti, talassemia e anemia calciforme sono causate da mutazioni genetiche nella sintesi dell’emoglobina, la proteina dei globuli rossi che trasporta ossigeno nell’organismo.
Nelle persone sane, ogni molecola di emoglobina è formata da 4 catene proteiche: 2 catene alfa e 2 catene beta. Nella talassemia e nell’anemia calciforme sono presenti delle alterazioni nelle catene proteiche.
Nella talassemia il problema è nelle catene beta, che nelle forme più gravi della malattia sono assenti o molto ridotte, mentre le catene alfa sono in eccesso. Questo squilibrio richiede periodiche trasfusioni di sangue.
Nell’anemia calciforme l’alterazione delle catene beta dell’emoglobina provoca la formazione di globuli rossi anomali, a forma di falce, da qui il nome della malattia. I globuli rossi deformati ostacolano il flusso sanguigno e l’ossigenazione nei capillari, provocando crisi molto dolorose e infarti nei tessuti.
La soluzione a questi problemi sta nell’emoglobina fetale, che non è formata da catene alfa-beta ma da catene alfa-gamma. Si tratta dell’emoglobina presente nel feto, che dopo la nascita viene progressivamente sostituita con quella composta da catene alfa-beta, per l’intervento del gene BCL11A, che blocca la sintesi delle catene gamma e attiva la produzione di catene beta. Questo meccanismo, tuttavia, non avviene sempre. Ci sono alcune persone, infatti, che continuano a produrre emoglobina fetale, con catene alfa-gamma, per tutta la vita. Questa condizione si chiama persistenza ereditaria di emoglobina fetale e non comporta problemi di salute.
Quando l’emoglobina fetale viene prodotta da persone con talassemia o anemia falciforme, le malattie hanno effetti molto più attenuati.
Il trattamento con editing del genoma consiste proprio nel ripristinare la sintesi dell’emoglobina fetale nei pazienti con talassemia e anemia calciforme.
Dai pazienti vengono prelevate cellule staminali emopoietiche che successivamente vengono modificate in appositi laboratori con il sistema CRISPR-Cas9, che viene programmato per “spegnere” il gene BCL11A e in questo modo far ripartire la produzione di emoglobina fetale. Dopo questa manipolazione genetica, le cellule modificate vengono infuse nei pazienti che nel frattempo sono stati sottoposti a una terapia farmacologica per “distruggere” il midollo, in modo da fare spazio alle nuove cellule staminali ingegnerizzate che si moltiplicheranno correggendo la malattia.
Un sistema estremamente sofisticato che sta dando risultati promettenti, presentati al 62° Congresso della Società Americana di Ematologia (ASH). Un appuntamento annuale che riunisce i maggiori esperti mondiali nel campo dell’ematologia e raccoglie i contributi scientifici più qualificati e innovativi al mondo nell’ambito delle malattie del sangue. Il congresso quest’anno si è svolto in via telematica.
Inoltre, i dati relativi ai primi 2 pazienti inseriti nello studio, uno affetto da talassemia trasfusione-dipendente e l’altro da anemia a cellule falciformi, sono stati pubblicati sull’autorevole rivista scientifica New England Journal of Medicine.
Grande soddisfazione è stata espressa dal prof. Locatelli per i risultati ottenuti nell’applicazione dell’editing genetico al primo paziente italiano. La sperimentazione prosegue e i risultati dovranno essere “verificati e confermati nel tempo”, ma l’inizio è molto promettente. Come ha sottolineato il professore, poi, “in futuro l’editing potrà essere utilizzato anche per il trattamento di altre malattie genetiche e per migliorare ulteriormente l’efficacia delle cellule CAR-T“.
Ulteriori informazioni sul sito web del Bambino Gesù.

LEGGI ANCHE: LEUCEMIA NEI BAMBINI: L’INCREDIBILE SUCCESSO DI UNA TECNICA DI TRAPIANTO
Che ne pensate di questa sperimentazione unimamme?